multiplex wormhole
In silico multiplex PCR primer design for noninvasive wildlife genetics.
![]()
Contents
- Description
- Installation
- Input File Format
- Quick Start: Panel design with the
multiplexWormholewrapper function - Multiplex Wormhole Functions:
- Batch design primers with batchPrimer3Design
- Tabulate predicted dimers with tabulateDimers
- Explore optimization parameters with plotASAtemps
- Optimize multiplex PCR primers with optimizeMultiplex
- Run multiple optimizations with multipleOptimizations
- Check specificity with primerTree_specificity_checks.R & offtargetThermodynamics
- Panel assessment wrapper function: assessPanel
- Recommended Workflows
- Handling Outputs
- Contact
- Success Stories
- Citations
Description
multiplex wormhole is largely an automation of pre-existing protocols for noninvasive SNP panel development (Eriksson et al. 2020). All default parameters for primer design, PCR parameters, and dimer prediction are based on this protocol and assume that the two-step PCR with Illumina Nextera adapters is being used. We recommend that the indexing step use Illumina unique dual indexes to minimize error rates due to index hopping, which is likely to be especially problematic with low-quality samples.
The heart of multiplex wormhole’s contribution to multiplex PCR primer design is its optimization algorithms. The multiplex wormhole optimize_multiplex.py function uses a combination of greedy, simple iterative improvement, and simulated annealing algorithms to minimize dimer load in the multiplex primer set. Dimer load can be measured by either: a) tallying pairwise dimers, or b) maximizing mean Gibbs free energy (deltaG; a meeasure of dimer strength) of pairwise dimers. The optimization process heavily relies on having abundant candidates relative to the number of target loci, and more templates needed for designing larger panels. See Optimization Details for a full descriptions of the optimization algorithms and parameter settings.
Installation
Set up a virtual environment
multiplex_wormhole was built and tested on MacOS with Python v3.9.13 in the Spyder IDE managed under Anaconda-Navigator. For those new to Python or with existing python packages, Anaconda is the recommended virtual environment manager. For a conda virtual environment within your working directory:
conda create -n py39 python=3.9 #create new virtual env w/ python v3.9
conda activate py39 #enter virtual env
The Oregon State University cluster uses pixi instead of conda for virtual environments:
pixi init #initialize virtual env
pixi add "python=3.9" #set python version
pixi add pip
pixi shell #enter virtual env
Install multiplex wormhole
pip install multiplex_wormhole
Note: Pixi/conda can be finicky… Dependening on your system, you may run into dependency errors here. If that happens, exit your virtual env and install the missing dependencies following the instructions below.
Back-up installation option
If pip install doesn’t work, you can also install manually by taking the following steps (from the command line):
# 1. Clone GitHub repository
git clone https://github.com/mhallerud/multiplex_wormhole/
# 2. Install python dependencies
pip install -r multiplex_wormhole/requirements.txt
# 3. Install multiplex_wormhole as python package
pip install -e multiplex_wormhole
# 4. Test install:
multiplex-wormhole -h
If the above steps fail, you can run the stand-alone scripts, e.g.:
cd multiplex_wormhole/src/multiplex_wormhole
python3.9 multiplexWormhole.py -h
Configuring the MFE primer binary
MFEprimer is used for dimer calculations. Multiplex wormhole is set up to automatically download and configure the binary file using the helpers/setup_mfeprimer.py script, take the following steps: Download the MFEprimer v3.2.7 release that fits your operating system here. Save the file to your multiplex_wormhole package directory (location can be found by running pip show multiplex_wormhole). If you cloned the repository directly from GitHub, save MFEprimer to multiplex_wormhole/src/multiplex_wormhole. Unzip the download (if zipped). Ensure the file can be executed by opening terminal or the command line in this directory and running chmod +x mfeprimer*.
Now you are ready to run multiplex wormhole!
Input file format
Templates file
The primary input to multiplex wormhole is a CSV table containing information on template DNA sequences and target regions. multiplex wormhole was developed for SNP-based genotyping with data originating from new or previously published genomic sequencing data or pre-existing SNP panels, but the method can be easily adjusted to accommodate multiplex PCR design for other targeted sequencing applications such as microsatellites, methylation regions, or functional regions.
Each DNA sequence in the CSV will be treated as a unique target for PCR amplification (i.e., sequences should be non-overlapping and unique). The file should include these three columns (exact capitalized field names required):
- SEQUENCE_ID : Template names, without punctuation. All punctuation will be automatically removed. Names should also be unique.
- SEQUENCE_TEMPLATE : DNA template sequence in the 5’–>3’ direction.
- SEQUENCE_TARGET : identifies the base pairs targeted for PCR ampflication following primer3 format: <startBP,length>. For example, a SNP at the 100th base pair in the sequence would be denoted as 100,1 in this field.
Example for defining the target field based on a DNA sequence with two target SNPs (red) at the 25th and 44th base pairs, respectively. The target length is the number of base pairs (inclusive) of both SNPs (i.e., the # of characters from 25 to 44 = 44-(25-1):
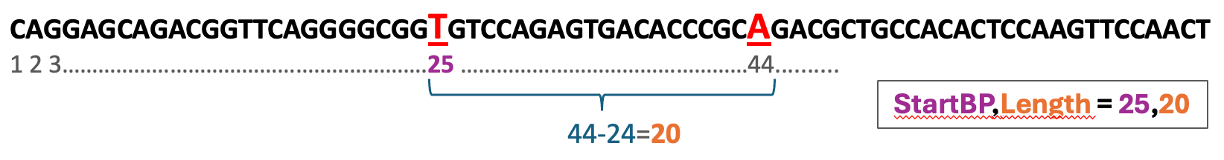
Example CSV format:
| SEQUENCE_ID | SEQUENCE_TEMPLATE | SEQUENCE_TARGET |
|---|---|---|
| CLocus_704 | TCAGAGAC… | 53,1 |
| … | … | … |
See the example input CSV.
The create_in_templates R script can be used to create this CSV from VCF and FASTA inputs. R dependencies include vcfR for VCF handling and openPrimeR for defining primer binding regions. The R script handles two input types:
-
de novo: Matches formatting output of de novo Stacks pipeline with FASTA created by
populations --fasta-loci. Assumes the VCF ‘CHROM’ field matches the FASTA sequence headers with a set prefix such as “CLocus_”. -
*reference-aligned: Assumes the FASTA sequence names are in the format
CHROM:startBP-endBPwhereCHROMmatches the reference sequence denoted as ‘CHROM’ field in the VCF,startBPis the position in the reference sequence where the FASTA sequence starts, andendBPis the position in the reference sequence where the FASTA sequence ends. SNPs will then be located within the FASTA sequences based on whether their position (POSin the VCF) is between startBP and endBP. This format can be extracted from reference-aligned SNP data in VCF format (e.g., output from Stacks, bcftools mpileup, etc.) using the following commands with REF.FASTA as the reference genome:
# NOTE: Flanking regions of 100-bp are used here as this is roughly the amplicon length targeted and provides good primer binding space on either side of the target SNP, but this can be adjusted if longer or shorter amplicons are desired.
# thin to 1 SNP per 100-bp to avoid overlapping template sequences
# this step is not necessary if linkage disequilibrium pruning has already been performed on the input VCF
vcftools --vcf <IN.VCF> --thin 100 --out Thinned100bp --recode
# convert SNPs into bed format
bcftools query -f '%CHROM\t%POS\n' Thinned100bp.recode.vcf | awk '{print $1"\t"$2-1"\t"$2}' | awk '{print $0"\t"$1":"$3}' > Thinned100bp.bed
# make tab-delimited file with reference sequence IDs in the first field and sequence lengths in the second field
seqkit fx2tab --length --name <REF.FASTA> chr_lengths.txt
# grab 100-bp flanking regions around thinned SNPs
bedtools slop -b 100 -i Thinned100bp.bed -g chr_lengths.txt > Thinned100bp_Flanking100bp.bed
# make fasta of these
bedtools getfasta -fi <REF.FASTA> -bed Thinned100bp_Flanking100bp.bed -fo <OUT.FA>
Primer and Keeplist files
Users can run multiplex wormhole steps with previously designed primers and can optionally include a “keeplist” FASTA file of primers which must be included in the final multiplex. In both files, primer names should follow the format
A Note on Candidate Loci
Multiplex wormhole treats all candidates equally, it is therefore the responsibility of the user to select informative candidate DNA sequences based on their objectives.
For example, I use the following steps to prepare input data for applications focused on individual identification:
- SNP discovery: Identifying SNPs from reduced representation sequencing (e.g., RADseq) or whole genome sequencing (WGS) projects, if available. Alternatively, SNP data can be pulled from templates of pre-existing panels, including Fluidigm and other SNP genotyping approaches.
- Initial SNP filtering: If using genomic data, follow standard genomics guidelines to remove erroneous SNPs (e.g., removing SNPs with low quality scores, low depths or extremely high depths, high missingness, etc.; O’Leary et al. 2018).
- Candidate SNP identification: SNPs with high information content based on the objective(s) of the panel are selected. For example, SNPs with high minor allele frequencies are informative for individual identification, while SNPs with high Fst or delta values are informative for population assignment.
- Extract flanking regions around SNPs: Using
populations --fasta-locifor de novo RADseq data andbedtools getfastafor reference-aligned RADseq or WGS data. For reference-aligned data, masking repetitive regions (e.g., using RepeatMasker) is recommended prior to this step. - Remove candidates in known repetitive regions: Check alignments against known repetitive elements using CENSOR. This step is recommended even with previous masking as repetitive regions / paralogs can cause genotyping error, particularly in low-quality samples where dropout rates are high.
Quick Start
The multiplexWormhole function is a wrapper around all sub-modules and will optimize a panel using the defaults for all other functions. Check out the full workflow and settings within individual functions for ultimate flexibility.
Command line usage
# PANEL DESIGN
# usage: multiplex-wormhole [-h] -t TEMPLATES -n NLOCI -o OUTDIR [-p PREFIX]
# [-k KEEPLIST] [-r RUNS] [-i ITER] [-c CYCLES]
# [-s SIMPLE] [-d] [-v]
# example for standard optimization with defaults:
python3.9 multiplexWormhole.py -t "Input_Templates.csv" -n 20 -o "Test_MW" -p "Test_MW_default" -k "Keeplist.fa"
# PANEL ASSESSMENT
# usage: mw-assess-panel [-h] -i INPUT [-a ALLDIMERS_DG] [-e ENDDIMERS_DG] [-b BADDIMERS_DG]
# example with defaults:
mw-assess-panel -i "Primers.fasta" -a -8 -e -5 -b -10
Python usage
# load module
import multiplex_wormhole as mw
# panel design example (showing defaults)
mw.multiplexWormhole(TEMPLATES="Input_Templates.csv",
N_LOCI=50,
OUTDIR="Test_MW",
PREFIX="Test_MW_default",
KEEPLIST_FA="Keeplist.fa",
N_RUNS=10, ITERATIONS=1000, CYCLES=10, SIMPLE=5000, deltaG=False, VERBOSE=False)#optional
# panel assessment example (showing defaults)
mw.assessPanel(PRIMERS,
ALL_DIMERS_dG=-8, END_DIMERS_dG=-4, BAD_DIMERS_dG=-10) #optional
Arguments
multiplexWormhole
TEMPLATES (-t –templates) : Path to templates CSV. NLOCI (-n –nloci) : Final panel size (i.e., # primer pairs & # templates amplified). OUTDIR (-o –outdir) : Filepath where output directory will be created and all outputs saved within a generated folder structure. PREFIX (-p –prefix) : Prefix for all outputs. [Defaults to a timestamp if None provided] KEEPLIST_FA (-k –keeplist) : Path to keeplist FASTA. [Default: None] N_RUNS (-r –runs) : Number of optimization runs. [Default: 10] ITERATIONS (-i –iter) : Number of simulated annealing iterations per cycle. [Default: 1000] CYCLES (-c –cycles) ** : Number of simulated annealing cycles per run. [Default: 10] **SIMPLE (-s –simple) : Number of simple iterative improvement iterations per run. [Default: 5000] deltaG (-d –deltaG) : Optimize for mean overall deltaG of dimers [True] or total dimer tally [False]? [Default: False] VERBOSE (-v –verbose) : Print all steps and swaps at the optimization step. [Default: False]
assessPanel
PRIMERS (-i –input) : FASTA or CSV of primers. Sequence names must match the format
multiplex-wormhole is a wrapper around the steps described below and uses their defaults. See the multiplex_wormhole page to understand the pipeline steps and output structure.
Multiplex Wormhole Functions
Click on the functions below for detailed information on inputs, outputs, and settings (including defaults).
-
Batch Primer Design with
mw-primer-design -
Dimer Prediction with
MFEprimer dimer -
Tabulate Dimers with
mw-tabulate-dimers(Optional): Explore Optimization Parameters with
mw-plot-simanneal -
Optimize Multiplex Primers with
mw-optimize_multiplex -
Multiple Run Optimization with
mw-mult_optimizations -
Panel Assessment with
mw-assess-panel -
Specificity Checks with
primerTree_specificity_checks.R&mw-specificity
Helper Functions:
- Convert CSV to FASTA with
mw-csv2fasta - Add Keeplist to FASTA with
mw-add-keeplist
See multiplex_primer_design.py for a full line-by-line workflow (equivalent to the multiplex-wormhole wrapper function).
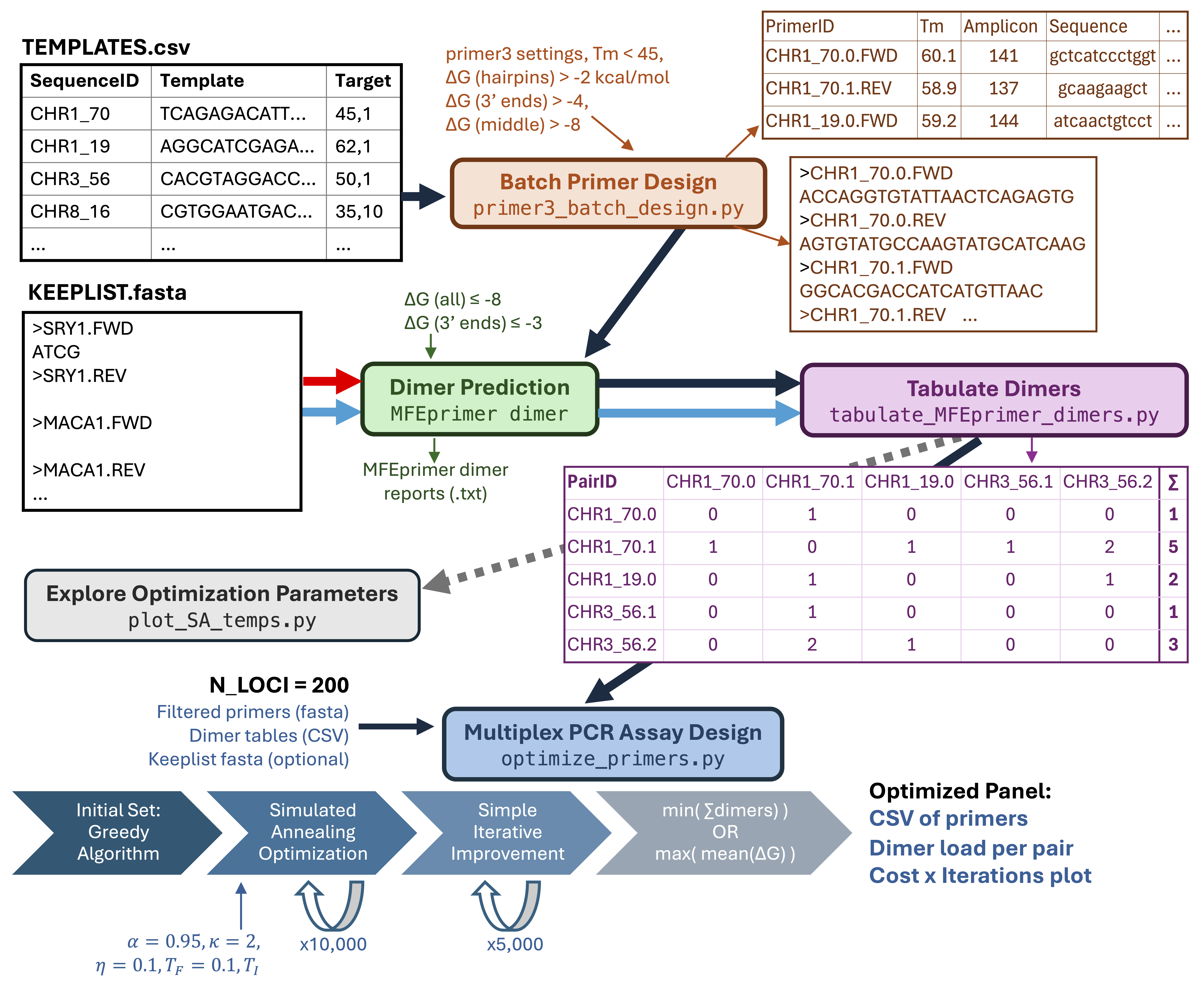
Diagram for multiplex wormhole workflow, with black arrows showing the panel design workflow and blue arrows showing the panel assessment workflow.
Recommended Workflows
Designing a Novel Panel
This workflow will optimize a novel panel from data including the full template DNA sequences (i.e., target + flanking regions).
TEMPLATES –> multiplex-wormhole
Augmenting an Existing Panel
This workflow will optimally expand an existing multiplex primer set (KEEPLIST) using new DNA template data (TEMPLATES).
TEMPLATES + KEEPLIST –> multiplex-wormhole
Assessing an Existing Panel
This workflow assesses dimer load of an existing multiplex primer set (PRMERS).
PRIMERS –> mw-assess-panel
Re-designing an Existing Panel
An existing panel can be redesigned, or multiple existing panels combined, but the approach will depend on whether only primer sequences available or the full templates are available. If template data are available, ensure that there is one template per target and use the “novel panel” or “augmented panel” (if unequal data availability) workflows. If only primer sequences are available, the only approach for optimization is to reduce panel size. To do so, use the following workflow with the original PRIMERS as the input fasta at the optimization step and set N_LOCI smaller than the current panel:
PRIMERS –> MFEprimer dimer –> mw-tabulate-dimers –> mw-mult-optimizations
Handling Outputs
Multiplex wormhole is an in silico design tool, and although it facilitates the primer design process, additional checks and lab optimization remain critical to panel success.
Dimer Checks
Dimer prediction is an imperfect science, and primers should be double-checked for particularly bad dimers e.g. by rerunning with MFEprimer v3 and/or other software such as MFEprimer v4, primer-dimer.com, and/or ThermoFisher’s Multiple Primer Analyzer.
Lab Testing
We recommend the protocol in Eriksson et al. (2020) for lab testing:
- Initial lab test in equimolar multiplex (0.2 uM per primer) on a range of sample qualities. Primer pairs that fail to amplify at this step and/or primers forming strong secondary structures that consume sequencing depth should be removed. Primer concentrations can also be adjusted to improve evenness of sequencing depth across targets.
- The second round of optimization should test specificity and genotyping error rates. These can be assessed using matched samples from the same individual (e.g., scat, hair, and tissue from the same animal) and testing on off-target species (e.g., diet items and closely related co-occurring species). Note that technical replicates are better than nothing, but are insufficient for assessing specificity and error issues specific to low-quality samples.
Problems? Questions?
Problems/bugs, questions, or ideas for enhancement can be directed to the GitHub Issues page. You can also directly contact Maggie Hallerud (hallerum@oregonstate.edu).
Success Stories
Multiplex wormhole has been used to develop or improve noninvasive genotyping panels for:







Contact us if you want to add your species to the list!
Citations
Eriksson, CE, Ruprecht J, Levi T. 2020. More affordable and effective noninvasive SNP genotyping using high-throughput amplicon sequencing. Molecular Ecology Resources 20(6): 1505:1516. doi: 10.1111/1755-0998.13208.
O’Leary, SJ, JB Puritz, SC Willis, CM Hollenbeck, and DS Portnoy. 2019. These aren’t the loci you’re looking for: Principles of effective SNP filtering for molecular ecologists. Molecular Ecology Resources 27(16): 3193-3206. doi: 10.1111/mec.14792.
Untergasser, A, Cutcutache, I, Koressaar, T, Ye, J, Faircloth, BC, Remm, M, Rozen SG. 2012. Primer3–new capabilities and interfaces. Nucleic Acids Research: e115. doi: 10.1093/nar/gks596.
Wang, K, H Li, Y Xu, Q Shao, J Yi, R Wang, W Cai, X Hang, C Zhang, H Cai, and W Qu. 2019. MFEprimer-3.0: quality control for PCR primers. Nucleic Acids Research 47 (W1): W610-W613. doi: 10.1093/nar/gkz351.