multiplex wormhole
Specificity Checking with PrimerTree
Ensuring primer specificity is an important and oft-neglected step towards successful and accurate genotyping of low-quality samples. This is particularly important when genotyping predator scats which can contain DNA from closely related species. Multiplex wormhole does not include a specificity check within the Python package, however, I’m providing an R script for post-processing multiplex wormhole outputs that cross-walks with the PrimerTree R package, which provides automated calling of PRIMER-BLAST.
Recommendations
- Run specificity checks against:
- The target (i.e., by checking against the target species or a close relative) to ensure that only one target in the genome is amplified. This is particularly important in noninvasive samples because sequence variation due to paralogs (i.e., regions present multiple times across the genome) is not differentiable from sequence dropout (e.g., due to variable degradation among loci), which is the most common form of genotyping error. Paralogs in highly variable regions (e.g., mRNA transcript variants) can be particularly problematic because dropout can be caused by variation in the primer binding region, causing biased amplification failure.
- Diet items to ensure that primers don’t amplify non-target DNA within scats, especially in predators who tend to consume prey within their same class. More closely related diet items are more likely to amplify.
- Potential overmarkers or common visitors to the same device setup (e.g., cooccurring carnivores may visit the same hair snare).
- Check against Bacteria if analyzing scats, since bacterial DNA will be abundant in scat samples.
- Check against human and other common lab contaminants.
- GenBank is a growing resource, and not all off-target species have good representation. I recommend using less stringent searches (e.g., family-level) rather than species-level searches.
- Off-target amplification can be ok (though never ideal), as long as the sequence is differentiable from the target amplicon (i.e. 2+ bp differences). Any off-target amplification within a 2-bp distance should be discarded because the resulting sequence variation in amplicons could be mistaken for SNPs.
Description
The PrimerTree_specificity_checks.R script provides a cross-walk between multiplex wormhole outputs and the PrimerTree R package (Cannon et al. 2016). PrimerTree enables automated calls to NCBI’s PRIMER-BLAST, which runs BLAST local alignments of primer pairs against specified taxonomic groups. PRIMER-BLAST uses modified BLAST settings to return results associated with lower matches that may still be amplified within PCR, and additionally applies a global Needleman-Wunsch alignment to ensure specificity, filters returned sequences dynamically based on the position of mismatches within the primer-binding regions, and filters sequnces for amplicon size (Untergasser et al. 2012). The provided R script: 1) automates running PRIMER-BLAST on each primer pair for a list of target taxa and outputs a CSV table, 2) extracts amplicon sequences and primer information for the final multiplex from multiplex wormhole inputs/intermediates, and 3) combines outputs from 1 and 2 via a global alignment, and plots a phylogenetic reconstruction including both the target amplicon sequences and the off-target sequences for each primer pair, with the number of basepair mismatches overlaid (Example below). An additional Python sub-module, offtarget_thermodynamics.py, calculates deltaG and Tm between each primer and off-target primer-binding site predicted by Primer-BLAST. DeltaG from these results can be used to filter off-target sequences included in plotting.
R Package Dependencies
For primerTree specificity checks:
- PrimerTree (Cannon et al. 2016)
- plyr (Wickham 2011)
- dplyr (Wickham et al. 2023) For phylogenetic reconstruction of alignments:
- DECIPHER (Wright 2016)
- Biostrings (Pagès et al. 2024)
Usage
Within R:
load("~/multiplex_wormhole/primerTree_specificity_checks.R")
1. Check Specificity with runPrimerTree (in R)
This function loops through each primer pair and organism combination and runs PRIMER-BLAST using primerTree::primer_search, merging results into a single dataframe. Taxonomy and sequences are then pulled from NCBI for each record using the primerTree::get_taxonomy and primerTree::get_sequence commands. The output is a dataframe containing the query primer pair (FWD & REV) and full PRIMER-BLAST outputs. Details on full functionality of primerTree.
library(primerTree)
library(plyr)
library(dplyr)
primerblast <- runPrimerTree(primers, organisms=c(),
fwd_adapter="tcgtcggcagcgtcagatgtgtataagagacag",
rev_adapter="gtctcgtgggctcggagatgtgtataagagacag",
all_combos=FALSE,
primer_specificity_database="core_nt",
exclude_env="False",
MAX_TARGET_SIZE=1000 #max amplicon size
...)
# check output
View(primerblast)
Arguments
primers : CSV output by multiplex wormhole optimization step, e.g., {OUTDIR}/3_OptimizedMultiplexes/Final_Primers/{OUTNAME}_Run01_primers.csv
organisms : Vector or list of organisms to search using GenBank taxid names. For example, to check specificity of marten primers within martens, co-occurring carnivores, diet items, and possible scat/lab contaminants, this list could include: c(“Mustelidae”, “Carnivora”, “Rodents and rabbits”, “Aves”, “Plethodontidae”, “Ericaceae”, “Bacteria”, “Homo sapiens”). Broader groups are preferred to specific diet items because many species are unlikely to have full genomes represented within GenBank. These names can be explored using the “Add Organism” button on the PRIMER-BLAST website.
all_combos : PRIMER-BLAST all FWD/REV combinations (NOTE: This will take awhile!).
primer_specificity_database : Search database (options=”core_nt”, “nt”, “PRIMERDB/genome_selected_species”, “refseq_mrna”, “refseq_representative_genomes”, “Custom”). If “Custom”, provide sequences in FASTA format to the “CUSTOM_DB” option.
exclude_env : Exclude NCBI sequences from environmental or uncultured samples?
MAX_TARGET_SIZE : Max amplicon size to return from PRIMER-BLAST.
… : Additional arguments that will be passed to primerTree::primer_search. These may include: num_aligns=500: # alignments to keep, .parallel=c(True,False) to enable parallel processing with foreach, .progress=c("none", "tk", "text") to enable a progress bar, or the full scope of PRIMER-BLAST options. If >500 returns are provided for a given taxonomic group + primer pair combination, rerun with increased num_aligns argument.
2. Calculate Thermodynamics of Off-target Primer-Binding Sites (in Python)
This function uses primer3-py to calculate thermodynamic stability of full binding sites and end stability of binding sites. Binding sites are identified from PrimerTree/PRIMER-BLAST outputs.
mw-specificity -i INFILE -o OUTFILE [-t ANNEAL_TEMP] [-m MV_CONC]
[-d DV_CONC] [-p DNTP_CONC] [-c DNA_CONC]
mw.offtargetThermodynamics(INFILE, OUTFILE, ANNEAL_TEMP=52.0, MV_CONC=50, DV_CONC=3.8, DNTP_CONC=0.25, DNA_CONC=50)
Arguments
INFILE (-i) : CSV containing PRIMER-BLAST outputs. Must contain ‘forward_start’, ‘forward_stop’, ‘reverse_start’, ‘reverse_stop’, ‘Sequence’, ‘FWDseq’, and ‘REVseq’ fields.
OUTFILE (-o) : Filepath where CSV results will save.
ANNEAL_TEMP (-t) : PCR annealing temp (Celsius). Default: 52
MV_CONC (-m) : Monovalent salt concentration in PCR. Default: 50
DV_CONC (-d) : Divalent salt concentration in PCR. Default: 3.8
DNTP_CONC (-p) : Primer concentration in PCR. Default: 0.25
DNA_CONC (-c) : DNA concentration in PCR. Default: 50
3. Extract Target Amplicons and Primer Information with extractTemplateInfo
This function extracts the amplicon sequence and primer information for an optimized multiplex based on the input, intermediate, and output files from multiplex wormhole. The output is a dataframe containing the primer information, template sequences and targets, and amplicon sequences and targets (i.e., the shifted target position relative to the full template).
primerinfo <- extractPrimerInfo(templates, filtprimers, finalprimers,
fwd_adapter="tcgtcggcagcgtcagatgtgtataagagacag", rev_adapter="gtctcgtgggctcggagatgtgtataagagacag")
View(primerinfo)
Arguments
templates : Input CSV containing template DNA sequences and targets input to multiplex wormhole (e.g, 0_Inputs/*Templates.csv)
filtprimers : CSV output by the primer3BatchDesign of multiplex wormhole (e.g., 1_PrimerDesign/FilteredPrimers.csv)
finalprimers : CSV of primers in optimized multiplex (e.g., 3_Optimized_Multiplexes/Final_Primers/{OUTNAME)_Run01_primers.csv)
fwd_adapter & rev_adapter : FWD & REV primer adapter sequences (5’–>3’), default is Illumina Nextera i5 & i7.
4. Align & Plot Specificity Check Results
This function uses the DECIPHER package’s maximum-likelihood trees with ancestral state reconstruction to visualize the number of mismatches between the target amplicon and sequences produced by in silico off-target amplification. For each primer pair, the target amplicon is aligned to off-target sequences with DECIPHER::DECIPHER::AlignSeqs, then a maximum likelihood dendrogram is constructed with DECIPHER::TreeLine(method=”ML”, reconstruct=True). TreeLine infers ancestral sequences for each node in the tree, then the number of mismatches can be calculated at each split. These state transitions are plotted at each node of the dendrogram and represent the number of mismatches between sequences or clusters. A sequence tree plot is made for each primer pair. Off-target sequences can be optionally filtered by delta G values from mw.offtargetThermodynamics results. To be conservative, the default is to plot all off-target sequences with deltaG<0 of the full binding site.
library(DECIPHER)
library(Biostrings)
# save to a PDF since this will be a bunch of plots
pdf("PRIMERBLAST_Trees.pdf") #open PDF
plotPrimerBlast(primerblast, primerinfo, species="TARGET", dG=0, dG_end=NA)
dev.off() #close PDF
Arguments
primerblast : Table output by runPrimerTree. May use other BLAST-like specificity check results as long as the table contains “FWD”, “REV”, “Sequence”, “accession”, and “species” fields, with sequences trimmed at the primer binding sites.
primerinfo : Table output by extractPrimerInfo. May use other inputs as longs as they contain “LocusID” and “AmpliconSeq” fields.
species : Prefix for amplicon sequences in plot. Recommended to fully capitalize to facilitate identification of target sequences within plots.
dG : Upper DeltaG threshold to include primer-binding sites. Both the FWD and REV primer-binding site must meet this threshold for the sequence to be plotted. Use ‘NA’ to skip, or if thermodynamics haven’t been calculated. (Default: 0)
dG_end : Upper DeltaG threshold for 3’ ends to include primer-binding sites. Both the FWD and REV primer-binding sites must meet this threshold for the sequence to be plotted. (Default: NA)
Output
This example shows predicted off-target amplification of Mustela, with a 51-bp difference in the Mustela sequence compared to the target Martes caurina amplicon.
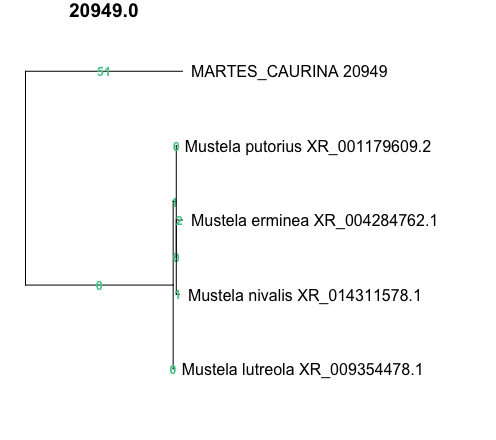
Citations
Cannon, M, j Hester, A Shalkhauser, ER Chan, K Logue, ST Small, D Serra. 2016. In silico assessment of primers for eDNA studies using PrimerTree and application to characterize the biodiversity surrounding the Cuyahoga River. Scientific Reports 6: 22908. doi:10.1038/srep22908
Pagès, H, P Aboyoun, R Gentleman, S DebRoy 2024. Biostrings: Efficient manipulation of biological strings. R package version 2.72.1, https://bioconductor.org/packages/Biostrings
Untergasser, A, I Cutcutache, T Koressaar, J Ye, BC Faircloth, M Remm, and SG Rozen. 2012. Primer3–new capabilities and interfaces. Nucleic Acids Research 40(15): e115. doi: 10.1093/nar/gks596
Wickham, H. 2011. The Split-Apply-Combine Strategy for Data Analysis. Journal of Statistical Software, 40(1), 1-29. https://www.jstatsoft.org/v40/i01/
Wickham, H, R François, L Henry, K Müller, D Vaughan. 2023. dplyr: A Grammar of Data Manipulation. R package version 1.1.4. https://CRAN.R-project.org/package=dplyr
Wright, ES. 2016. Using DECIPHER v2.0 to Analyze Big Biological Sequence Data in R. The R Journal: 8(1): 352-359. doi: 10.32614/RJ-2016-025
PRIMER-BLAST OPTIONS
This is a list of PRIMER-BLAST options that can be input as arguments into primerTree::primer_search. Defaults shown. To determine which option corresponds to which PRIMER-BLAST webpage setting, you can right-click on the setting on the website and select “Inspect element”, then look for the corresponding “name” and “value”.
- SEQFILE=NA,
- PRIMER5_START=NA,
- PRIMER5_END=NA,
- PRIMER3_START=NA,
- PRIMER3_END=NA,
- PRIMER_LEFT_INPUT=NA,
- PRIMER_RIGHT_INPUT=NA,
- PRIMER_PRODUCT_MIN=70,
- PRIMER_PRODUCT_MAX=1000,
- PRIMER_NUM_RETURN=10,
- PRIMER_MIN_TM=57.0,
- PRIMER_OPT_TM=60.0,
- PRIMER_MAX_TM=63.0,
- PRIMER_MAX_DIFF_TM=3,
- PREFER_3END=,
- PRIMER_ON_SPLICE_SITE=0,
- SPLICE_SITE_OVERLAP_5END=7,
- SPLICE_SITE_OVERLAP_3END=4,
- SPLICE_SITE_OVERLAP_3END_MAX=8,
- SPAN_INTRON=,
- MIN_INTRON_SIZE=1000,
- MAX_INTRON_SIZE=1000000,
- SEARCH_SPECIFIC_PRIMER=”on”,
- SEARCHMODE=0,
- PRIMER_SPECIFICITY_DATABASE=”nt”,
- CUSTOMSEQFILE=NA,
- EXCLUDE_XM=NA,
- EXCLUDE_ENV=NA,
- ORGANISM=”Homo sapiens”,
- ORGANISM2=”rodents and rabbits 314147”,
- slctOrg=,
- ENTREZ_QUERY=NA,
- TOTAL_PRIMER_SPECIFICITY_MISMATCH=1,
- PRIMER_3END_SPECIFICITY_MISMATCH=1,
- MISMATCH_REGION_LENGTH=5,
- TOTAL_MISMATCH_IGNORE=6,
- MAX_TARGET_SIZE=1000,
- ALLOW_TRANSCRIPT_VARIANTS=NA,
- NEWWIN=NA,
- SHOW_SVIEWER=on,
- HITSIZE=50000,
- EVALUE=30000,
- WORD_SIZE=7,
- MAX_CANDIDATE_PRIMER=500,
- NUM_TARGETS=20,
- NUM_TARGETS_WITH_PRIMERS=1000,
- MAX_TARGET_PER_TEMPLATE=100
- PRODUCT_MIN_TM=,
- PRODUCT_OPT_TM=,
- PRODUCT_MAX_TM=,
- PRIMER_MIN_SIZE=15,
- PRIMER_OPT_SIZE=20,
- PRIMER_MAX_SIZE=25,
- PRIMER_MIN_GC=20.0,
- PRIMER_MAX_GC=80.0,
- GC_CLAMP=0,
- POLYX=5,
- PRIMER_MAX_END_STABILITY=9,
- PRIMER_MAX_END_GC=5,
- TH_OLOGO_ALIGNMENT=,
- TH_TEMPLATE_ALIGNMENT=,
- PRIMER_MAX_TEMPLATE_MISPRIMING_TH=40.00,
- PRIMER_PAIR_MAX_TEMPLATE_MISPRIMING_TH=70.00,
- PRIMER_MAX_SELF_ANY_TH=45.0,
- PRIMER_MAX_SELF_END_TH=35.0,
- PRIMER_PAIR_MAX_COMPL_ANY_TH=45.0,
- PRIMER_PAIR_MAX_COMPL_END_TH=35.0,
- PRIMER_MAX_HAIRPIN_TH=24.0,
- PRIMER_MAX_TEMPLATE_MISPRIMING=12.00,
- PRIMER_PAIR_MAX_TEMPLATE_MISPRIMING=24.00,
- SELF_ANY=8.00,
- SELF_END=3.00,
- PRIMER_PAIR_MAX_COMPL_ANY=8.00,
- PRIMER_PAIR_MAX_COMPL_END=3.00,
- EXCLUDED_REGIONS=NA,
- OVERLAP=NA,
- OVERLAP_5END=7,
- OVERLAP_3END=4,
- MONO_CATIONS=50.0,
- DIVA_CATIONS=1.5,
- CON_DNTPS=0.6,
- SALT_FORMULAR=1,
- TM_METHOD=1,
- CON_ANEAL_OLIGO=50.0,
- NO_SNP=,
- PRIMER_MISPRIMING_LIBRARY=AUTO,
- LOW_COMPLEXITY_FILTER=on,
- PICK_HYB_PROBE=,
- PRIMER_INTERNAL_OLIGO_MIN_SIZE=18,
- PRIMER_INTERNAL_OLIGO_OPT_SIZE=20,
- PRIMER_INTERNAL_OLIGO_MAX_SIZE=27,
- PRIMER_INTERNAL_OLIGO_MIN_TM=57.0,
- PRIMER_INTERNAL_OLIGO_OPT_TM=60.0,
- PRIMER_INTERNAL_OLIGO_MAX_TM=63.0,
- PRIMER_INTERNAL_OLIGO_MIN_GC=20.0,
- PRIMER_INTERNAL_OLIGO_OPT_GC_PERCENT=50,
- PRIMER_INTERNAL_OLIGO_MAX_GC=80.0,
- NA=NA NEWWIN=, SHOW_SVIEWER=on